
成体干细胞在损伤后会迅速但短暂地被激活,以修复受损组织。反复或慢性损伤则会增加癌症风险。尽管人们在识别和表征哺乳动物干细胞以及调节其增殖的稳态促分裂原方面取得了进展,但损伤激活的促分裂原、使其在损伤前保持失活的机制,以及这些过程是否及如何促进癌症的发生,尚不清楚。一项最新研究针对小鼠肺部一种名为NEstem的上皮干细胞群体,回答了这些问题。

NEstem是稀有的干细胞,与其他肺神经内分泌细胞混合在一起,存在于受神经支配的神经感觉簇中。这些感觉簇监测气道状态,并充当干细胞的微环境。NEstem是兼性干细胞,在气道损伤后广泛增殖,形成大的克隆修复斑块,恢复周围上皮。NEstem也可通过联合缺失肿瘤抑制因子Rb和p53而被永久激活,从而启动小细胞肺癌(SCLC)——一种最致命的癌症。
在一项新的研究中,研究人员试图识别损伤诱导NEstem的促分裂原、损伤激活促分裂原并刺激干细胞增殖的机制,以及这一组织修复通路与SCLC肿瘤抑制因子的关系。他们认为,这可以揭示损伤如何触发干细胞激活(分裂)关键第一步的分子逻辑,并回答组织修复、干细胞和癌症领域的问题。
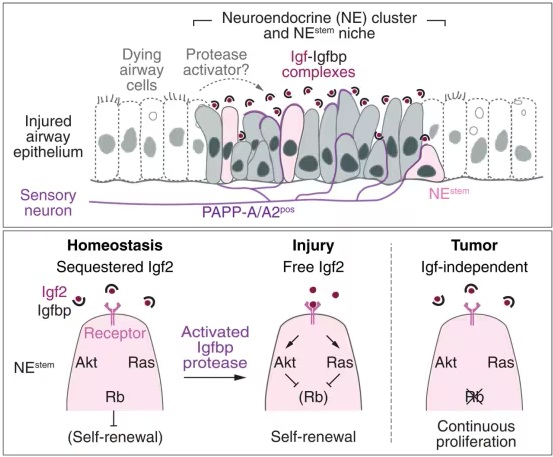
为了识别促分裂信号,研究人员开发了一种小鼠肺切片培养测定法,并筛选了候选配体对NEstem增殖的影响。只有胰岛素样生长因子1和2(Igf1和Igf2)能诱导其增殖。Igf2及其受体在损伤诱导的NEstem激活中既被表达,也是必需的,因此以自分泌方式发挥作用。尽管NEstem组成型产生Igf2,但它被共表达的Igf结合蛋白(Igfbps)以非活性形式隔离并稳定在微环境中。通过药物从潜伏的Igf-Igfbp复合物中释放Igf,或通过微环境中感觉神经元表达的PAPP-A和PAPP-A2蛋白酶破坏Igfbp,都能立即触发NEstem增殖,损伤也是如此。Igf信号传导抑制了通常强制干细胞静止的Rb功能。通过Rb缺失导致的持续通路激活引起了连续的干细胞增殖。
综上所述,这项新的研究表明,除了其经典的激素作用外,Igf还在肺干细胞微环境中以潜伏的自分泌复合物(与Igfbp结合)形式局部发挥作用,随时准备被损伤激活。损伤释放Igf,推测是通过PAPP-A和/或PAPP-A2对Igfbp的蛋白水解破坏。一旦释放,Igf激活其信号通路,抑制Rb并释放细胞周期检查点,从而启动干细胞增殖。这种组织修复中的“待命”控制通路可能在癌症中被利用,因为Rb缺失会立即启动不受控制的NEstem增殖和肿瘤发生。这种局部的、可快速激活的Igf-Rb干细胞控制通路也可能在身体和大脑的其他神经内分泌和神经干细胞及肿瘤中运作。
研究人员揭示了一个自我强化的前馈循环:HNF1B缺乏驱动CKD,而CKD相关应激则表观遗传抑制HNF1B活性。
慢性肾脏病(CKD)影响着全球超过10%的人口,其特征是肾功能不可逆转地持续下降,无论最初的损伤原因是什么。CKD的一个决定性特征是它的自我维持性:一旦疾病确立,即使最初的触发因素已经消失,肾损伤仍会继续进展。识别维持这种病理状态的分子回路,对于理解疾病进展和设计有效的治疗策略至关重要。
HNF1B是一种对肾脏发育至关重要的转录因子。HNF1B的杂合功能丧失突变会导致常染色体显性小管间质性肾病,这是一种罕见的单基因疾病,其肾脏组织病理学与常见CKD的表型相似。这种表型趋同表明,HNF1B功能障碍可能代表了连接罕见遗传性肾脏病与更常见的多因素CKD的一个共享机制。在一项新的研究中,研究人员假设,在CKD过程中,HNF1B活性可能被逐渐抑制,从而形成一个推动疾病进展的前馈循环。

使用小鼠模型,研究人员发现,出生后在肾小管上皮细胞中失活Hnf1b会导致非常快速和严重的CKD,其特征为肾小管萎缩、间质纤维化、炎症和进行性肾衰竭。Hnf1b的缺失破坏了上皮分化,并触发了正常静止的小管细胞突然重新进入细胞周期。这种非计划的增殖引起了复制应激、DNA损伤、凋亡和衰老,从而导致CKD。使用CDK4/6抑制剂palbociclib对异常细胞周期进入进行药理学抑制,略微减轻了Hnf1b缺陷小鼠的小管损伤和纤维化,表明复制应激参与了HNF1B缺失下游的疾病进展。
为了识别HNF1B缺失的最早分子后果,研究人员在肾损伤发生之前就确定了HNF1B的转录特征。在由独立于HNF1B的损伤(包括次全肾切除术、Alport综合征、缺血再灌注损伤、肾病综合征和单侧输尿管梗阻)引发的多种CKD临床前模型中,这一特征显著减少。值得注意的是,HNF1B活性的抑制发生在任何明显的组织学损伤出现之前,并且与急性肾损伤后上皮修复失败相关。
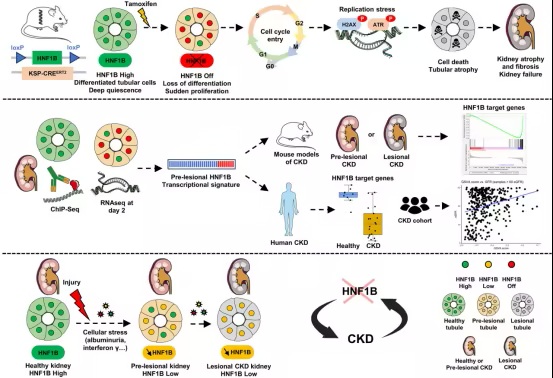
在机制上,研究人员发现,常见的CKD相关应激(包括蛋白尿和干扰素-γ)会降低肾小管细胞中HNF1B的转录活性。单细胞转录组和染色质可及性分析显示,受损和适应不良修复的小管细胞在HNF1B结合位点表现出可及性降低,将上皮应激与HNF1B功能的表观遗传抑制联系起来。
最后,对包括近900份活检样本(涵盖广泛CKD严重程度)的人类肾脏转录组数据集的分析显示,HNF1B靶基因表达降低与肾功能下降、肾小管萎缩和纤维化密切相关。这些发现确立了HNF1B活性作为人类CKD严重程度的分子决定因素。
综上所述,这些研究结果表明,HNF1B是肾小管稳态的守护者,其缺失启动并维持CKD。研究人员揭示了一个自我强化的前馈循环:HNF1B缺乏驱动CKD,而CKD相关应激则表观遗传抑制HNF1B活性。这一机制连接了罕见的单基因肾脏病和常见的CKD形式,并为理解CKD的无情本质提供了一个概念框架。恢复HNF1B活性可能代表一种改变CKD进程的治疗策略。
阿片类药物既是疼痛治疗的中流砥柱,也是成瘾与过量死亡的罪魁祸首;仅在美国,2023年就有超过8万人死于合成阿片类药物过量,其中绝大多数与芬太尼相关。长期以来,科学界形成了一条共识:μ-阿片受体高内在活性的激动剂虽然镇痛效果强大,但必然伴随呼吸抑制、耐受、依赖和成瘾等严重副作用。
然而,一篇发表在国际杂志Nature上题为“A μ opioid receptor superagonist analgesic with minimal adverse effects”的研究报告中,来自美国国立卫生研究院的研究人员等机构的科学家们通过研究用一种名为DFNZ的新型分子正面挑战了这一教条。
文章中,研究人员重新审视了一类被尘封数十年的合成苯并咪唑类阿片药物—硝氮烯类,这类化合物在1950年代因效力过强而被搁置,研究人员首先关注了其中一种名为FNZ的衍生物,并用正电子发射断层扫描成像技术在大鼠体内追踪它的行踪。结果发现,FNZ进入大脑后仅停留5到10分钟,但镇痛效果却持续了至少两个小时;这种“药物已走,效果还在”的矛盾现象提示,很可能存在某种活性代谢物在起作用。进一步的筛选最终锁定了目标:N-去乙基-氟硝氮烯,一种在μ-阿片受体上具有超高内在活性的“超级激动剂”。
DFNZ的药理学特征完全不同于传统阿片药物,在治疗剂量下,其不仅不抑制呼吸,反而适度且持续地增加脑组织氧水平。重复给药后,大鼠没有出现耐受、药物依赖性,也几乎没有戒断症状—在经典的14项阿片戒断指标中,研究人员仅观察到了处理动物时的易激惹表现。更令人惊讶的是,DFNZ不引起μ-阿片受体的下调,这一点与高功效激动剂的典型表现截然相反。
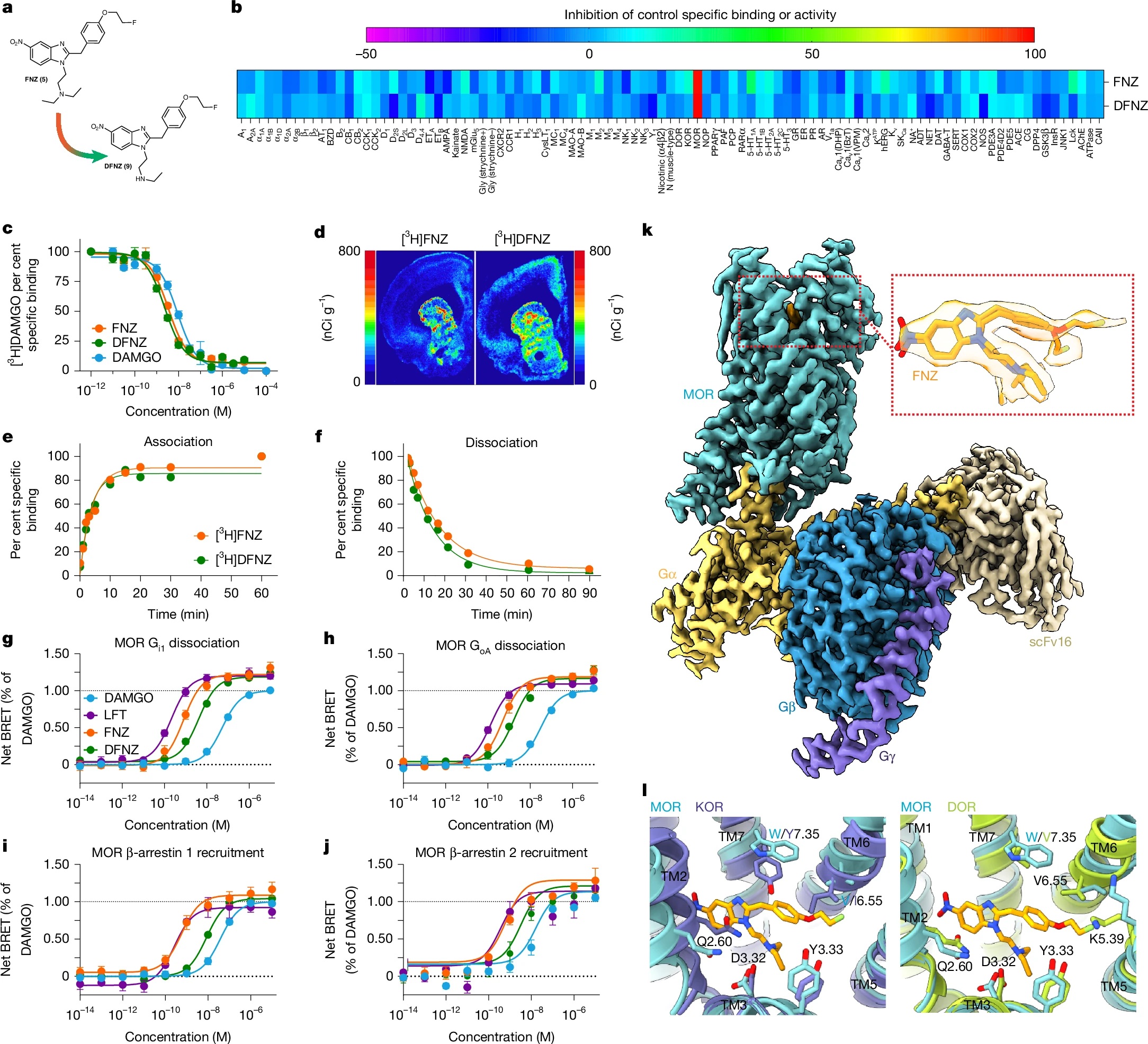
为了评估其成瘾潜力,研究人员测试了大鼠的自我给药行为;结果显示,动物确实会主动按压杠杆获取DFNZ,说明其具有一定的奖赏效应;但关键的区别在于:当药物被换成生理盐水后,动物立即停止了觅药行为,这一表现与海洛因、吗啡和芬太尼完全不同,后者的动物在药物移除后仍会持续寻求药物。神经化学层面的分析解释了这一现象:DFNZ能缓慢增加伏隔核中的多巴胺水平,但不会触发与强效药物-线索关联形成相关的快速多巴胺爆发;后者正是驱动渴求和复吸的条件性反应的基础。
那么,一个高效能的μ-阿片受体激动剂,如何做到“镇痛强、成瘾低”?机制研究揭示,DFNZ独特的时空细胞信号谱起了关键作用,其还在μ-阿片受体与甘丙肽1型受体的异源二聚体上表现出减弱的功效。这种独特的药理学轮廓,使其在某些情境下又表现出类似低内在效能部分激动剂的特征,而后者一直被认为是安全性所需的药理学属性。
这项研究的意义远不止发现一个新分子,其直接挑战了一个主导阿片药物研发数十年的核心假设:高功效μ-阿片受体激动剂不可能成为安全的治疗药物。事实上,研究人员认为,DFNZ甚至有望用于治疗阿片使用障碍,且可能优于现有的激动剂类药物,后者仍存在呼吸抑制风险。
当然,目前的研究仍处于临床前阶段,研究人员正在推进更多动物实验,从而为申请人体试验许可做好准备;对于手术疼痛、癌性疼痛及对有效镇痛需求极为迫切的慢性疼痛患者,这类“不走寻常路”的分子或许能带来真正的转机。
文章转载自生物谷,系出于传递更多信息之目的,转载内容不代表本站立场。如有侵权请及时联系,我们将立即进行删除处理。

 京公网安备 11011402010692号
京公网安备 11011402010692号





{kind=link}